Мальформация Киари: причины, симптомы, типы, диагностика, лечение
Что такое мальформация Киари?
Мальформация Киари - это структурные дефекты в основании черепа и мозжечка, части мозга, который контролирует баланс. Обычно мозжечок и части ствола мозга располагаются над затылочным отверстием в черепе, которое позволяет проходить сквозь него спинному мозгу (так называемый маятник). Когда часть мозжечка опускается ниже затылочного отверстия и в верхний спинномозговый канал, это называется мальформацией Киари.
Мальформация Киари может развиваться, когда часть черепа меньше нормальной величины или деформирована, что заставляет мозжечок опускаться вниз в затылочное отверстие и в позвоночный канал. Что вызывает давление на мозжечок и на продолговатый мозг, которое может оказывать негативное влияние на функции этих областей головного мозга, и блокировать поток спинномозговой жидкости - прозрачной жидкости, которая окружает и смягчает головной и спинной мозг. Спинномозговая жидкость также циркулирует питательные вещества и химические вещества, отфильтрованные из крови, и удаляет отходы из головного мозга.
Причины мальформации Киари
У мальформации Киари существует несколько причин. Чаще всего болезнь вызвана структурными дефектами головного и спинного мозга, которые возникают во время развития плода. Это может быть результатом генетических мутаций или неправильного питания матери, отсутствия определенных витаминов или питательных веществ. Эта форма называется первичной или врожденной мальформацией Киари.
Патология также может быть развиваться позже, если спинальная жидкость слишком сильно истощена от поясничной или грудной отделов позвоночника либо из-за травматического повреждения, болезни, либо инфекции. Эта форма называется приобретенной или вторичной мальформацией Киари. Первичная форма Киари гораздо более распространена, чем вторичная мальформация Киари.
Симптомы мальформации Киари
Головная боль является отличительным признаком мальформации Киари, особенно после внезапного кашля, чихания или напряжения. Другие симптомы могут различаться у больных и могут включать в себя:
- боль в шее
- проблемы со слухом или равновесием
- мышечная слабость или онемение
- головокружение
- трудности с глотанием или с речью
- рвота
- звон или жужжание в ушах (тиннитус)
- искривление позвоночника (сколиоз)
- бессонница
- депрессия
- проблемы с координацией рук и точных моторных навыков.
Некоторые люди с СМ могут не проявлять никаких симптомов. Симптомы могут изменяться для некоторых людей, в зависимости от сжатия ткани и нервов и от нарастания давления CSF.
Младенцы с мальформацией Киари могут испытывать трудности с глотанием, раздражительностью при кормлении, чрезмерным слюнотечением, слабым криком, рвотой или рвотой, слабостью руки, жесткой шеей, проблемами с дыханием, задержками развития и невозможностью набрать вес.
Типы мальформации Киари
Мальформации Киари классифицируются по степени тяжести расстройства и в зависимости от частей мозга, которые опускаются в спинномозговой канал.
Мальформация Киари Тип I
Тип 1 возникает, когда нижняя часть мозжечка (называемая мозжечковыми миндалинами) распространяется в затылочное отверстие. Обычно через это отверстие проходит только спинной мозг. Тип 1, который не вызывает симптомы, является наиболее распространенной формой мальформации Киари. Обычно заболевание устанавливается в подростковом возрасте или в зрелом возрасте, часто случайно при обследовании на другое заболевание.
Мальформация Киари Тип II
Люди, страдающие типом II имеют симптомы, которые обычно более тяжелые, чем у Типа 1, и обычно появляются в детстве. Это заболевание может вызвать опасные для жизни осложнения во время младенчества или раннего детства, и лечение этого требует хирургического вмешательства.
При II типе, также называемом классической мальформацией Киари, как мозжечок, так и продолговатый мозг опускаются в затылочное отверстие. Также нервная ткань, которая соединяет две половины мозжечка, может отсутствовать или быть только частично сформирована. Тип II обычно сопровождается миеломенсингоцеле - формой расщепления позвоночника, которая возникает, когда спинальный канал и позвоночник не закрываются до рождения. (Spina bifida - это расстройство, характеризующееся неполным развитием головного мозга, спинного мозга и / или их защитного покрытия.) Миеломинеоцеле обычно приводит к частичному или полному параличу области ниже спинного отверстия. Термин аномалия Арнольд-Киари (названный в честь двух новаторских исследователей) специфичен для мальформаций Типа II.
Мальформация Киари Тип III
Тип III очень редок и является наиболее серьезной формой мальформации Киари. При данном типе заболевания некоторая часть мозжечка и продолговатый мозг выступают через аномальное отверстие в задней части черепа. Это может также включать мембраны, окружающие мозг или спинной мозг.
Симптомы типа III проявляются в младенчестве и могут вызывать серьезные опасные для жизни осложнения. Младенцы, имеющие III тип болезни, могут иметь многие из тех же симптомов, что и у пациентов с типом II, но также могут иметь дополнительные серьезные неврологические дефекты, такие как умственные и физические задержки и судороги.
Мальформация Киари Тип IV
Тип IV характеризуется неполным или недоразвитым мозжечком (состояние, известное как гипоплазия мозжечка). В этой редкой форме заболевания мозжечок расположен в нормальном положении, но его части отсутствуют, а части черепа и спинного мозга могут быть видны.
Какие еще условия связаны с мальформацией Киари?
Гидроцефалия - это чрезмерное накопление спинномозговой жидкости в головном мозге. Мальформация Киари может блокировать нормальный поток этой жидкости и вызывать давление внутри головы, что может привести к умственным дефектам и / или увеличенному или деформируемому черепу. Тяжелая гидроцефалия, если ее не лечить, может привести к летальному исходу. Расстройство может происходить с любым типом пороков Киари, но чаще всего ассоциируется с типом II.
Расщепление позвоночника (Spina bifida) - это неполное закрытие позвоночника и мембран вокруг спинного мозга. У младенцев с расщелиной позвоночника кости вокруг спинного мозга не образуются должным образом, вызывая дефекты в нижнем отделе позвоночника. В то время как для большинства детей с этим врожденным дефектом свойственна умеренная форма, при которой неврологические проблемы отсутствуют, у людей с мальформацией типа II Киари обычно возникает миеломенсингоцеле, а спинной мозг ребенка остается открытым в одной области спины и нижнего отдела позвоночника. Мембраны и спинной мозг выступают через отверстие в позвоночнике, создавая мешок на спине ребенка. Это может вызвать ряд неврологических нарушений, таких как мышечная слабость, паралич и сколиоз.
Сирингомиелия - это расстройство, в котором заполненная трубчатая киста или сиринкс образуется в центральном канале спинного мозга. Растущий сиринкс разрушает центр спинного мозга, что приводит к боли, слабости и жесткости в спине, плечах, руках или ногах. Другие симптомы могут включать потерю способности чувствовать крайности горячего или холодного, особенно в руках. У некоторых людей также возникает сильная боль в груди и шее.
Синдром связанного корда возникает, когда спинной мозг ребенка аномально прикрепляется к тканям вокруг нижней части позвоночника. Это означает, что спинной мозг не может свободно перемещаться по спинному каналу. По мере роста ребенка расстройство ухудшается и может привести к постоянному повреждению нервов, которые контролируют мышцы нижнего тела и ног. У детей, у которых развивается миеломинеоцеле, существует повышенный риск развития привязанного шнура позже в жизни.
Спинная кривизна распространена среди лиц с сирингомиелией или первым типом мальформации Киари. Позвоночник может либо сгибаться влево, либо вправо (сколиоз) или может сгибаться вперед (кифоз).
Эпидемиология мальформации Киари
Раньше было подсчитано, что заболевание происходит примерно в одном на каждые 1000 рождений. Однако более широкое использование диагностической визуализации показало, что расстройство Киари может быть гораздо более распространенным явлением. Сложностью этих статистических исследований является тот факт, что у некоторых детей, родившихся с данной патологией, никогда не проявляются симптомы или проявляются только в подростковом возрасте или в зрелом возрасте. Мальформация Киари чаще встречаются у женщин, чем у мужчин, а пороки развития II типа более распространены в некоторых этнических группах, включая людей кельтского происхождения.
Диагностика мальформации Киари
В настоящее время не существует теста для определения родится ли ребенок с мальформацией Киари. Так как патология зависит от определенных врожденных нарушений, таких как расщепление позвоночника, дети, родившиеся с этими дефектами, часто испытывают пороки развития. Тем не менее, некоторые пороки развития можно обнаружить с помощью ультразвуковых изображений до рождения.
Многие люди с мальформацией Киари не испытывают симптомов, и их пороки развития обнаруживаются только в ходе диагностики или лечения другого заболевания. Врач выполняет физический осмотр и проверяет память, сознание, равновесие и баланс (функции, контролируемые мозжечком), осязание, рефлексы, ощущения и моторные навыки (функции, контролируемые спинным мозгом).
Врач может также назначить один из следующих диагностических тестов:
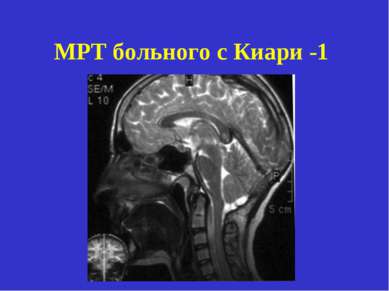
Магнитно-резонансная томография (МРТ) - это процедура визуализации, наиболее часто используемая для диагностики мальформации Киари. Она использует радиоволны и мощное магнитное поле, чтобы безболезненно производить либо подробную трехмерную картину, либо двумерный «срез» структур тела, включая ткани, органы, кости и нервы.
Рентгеновские лучи используют электромагнитную энергию для получения изображений костей и определенных тканей на пленке. Рентген головы и шеи не может выявить патологию, но может идентифицировать аномалии костей, которые часто связаны с заболеванием.
Компьютерная томография (КТ) использует рентгеновские лучи и компьютер для создания двумерных изображений костей и кровеносных сосудов. КТ может идентифицировать гидроцефалию и аномалии костей, связанные с мальформацией Киари.
Лечение мальформации Киари
Некоторые мальформации Киари не проявляют симптомов и не мешают повседневной жизни человека. В этих случаях врачи могут рекомендовать регулярный мониторинг с помощью МРТ. Когда люди испытывают боль или головную боль, врачи могут назначать лекарства для облегчения симптомов.
Хирургия
Во многих случаях хирургическое вмешательство является единственным средством для облегчения симптомов или прекращением прогрессирования повреждения центральной нервной системы. Хирургия может улучшить или стабилизировать симптомы у большинства людей. Для лечения состояния может потребоваться более одной операции.
Наиболее распространенной хирургической операцией для лечения мальформации Киари является декомпрессия задней ямки. Это создает больше пространства для мозжечка и снимает давление на спинной мозг. Операция включает в себя надрез в задней части головы и удаление небольшой части кости в нижней части черепа (краниэктомия). В некоторых случаях также может быть удалена арочная костная крыша позвоночного канала, называемая пластинкой (спинальная ламинэктомия). Операция должна помочь восстановить нормальный поток спинномозговой жидкости, и в некоторых случаях этого может быть достаточно, чтобы облегчить симптомы.
Затем хирург может сделать надрез в твердой мозговой оболочке (защитное покрытие головного и спинного мозга. Некоторые хирурги проводят допплеровский ультразвуковой тест во время операции, чтобы определить, требуется ли открытие оболочки. Если область мозга и спинного мозга все еще переполнена, хирург может использовать процедуру, называемую электрокаутерией, для удаления мозжечковых миндалин, что позволяет использовать больше свободного места. Эти миндалины не имеют какой-либо функции и могут быть удалены без каких-либо известных неврологических проблем.
Последний шаг - сшить паз твердой мозговой оболочки, чтобы расширить пространство вокруг миндалин, подобно выпуску пояса из штанов. Этот паз может быть сделан из искусственного материала или ткани, собранной из другой части тела человека.
Младенцам и детям с миеломенсингоцеле может потребоваться хирургическое вмешательство для перемещения спинного мозга и закрытия отверстия в спине. Эта операция наиболее эффективна, когда она проводится пренатально (пока ребенок все еще находится в утробе матери), а не после рождения. Пренатальная хирургия уменьшает появление гидроцефалии и восстанавливает мозжечок и мозговой шток до более нормального выравнивания.
Гидроцефалию можно лечить системой шунта (трубки), которая истощает избыток жидкости и снимает давление внутри головы. Прочная трубка, хирургически вставленная в головку, соединена с гибкой трубкой, помещенной под кожу. Эти трубки сливают избыток жидкости в полость грудной клетки или живот, чтобы он мог поглощаться телом.
Альтернативным хирургическим лечением у некоторых людей с гидроцефалией является третья вентрикулостомия, процедура, которая улучшает течение спинномозговой жидкости из головного мозга. Небольшое отверстие выполнено на дне третьего желудочка (полость мозга), и жидкость отводится для уменьшения давления. Аналогичным образом, в тех случаях, когда операция неэффективна, врачи могут открывать спинной мозг и вставлять шунт для слива сирингомиелии или гидромелии (повышенная жидкость в центральном канале спинного мозга).
ЛАБОРАТОРНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ДЕТЕЙ ПЕРВЫХ 3 ЛЕТ ЖИЗНИ В НЕВРОЛОГИЧЕСКОЙ КЛИНИКЕ
ЕЩЕ: НА ГЛАВНУЮ СТРАНИЦУ
Избранные статьи
