Болезнь Шарко-Мари-Тута: симптомы, причины, диагностика, лечение
Что такое болезнь Шарко-Мари-Тута?
Болезнь Шарко-Мари-Тута (Charcot-Marie-Tooth) является одним из наиболее распространенных наследственных неврологических заболеваний, которым страдает примерно 1 из 2500 человек в мире. Болезнь названа в честь трех врачей, которые впервые описали ее в 1886 году: Жан-Мартин Шарко и Пьер Мари в Париже, Франция, и Говард Генри Тут в Кембридже, Англия. Болезнь Шарко-Мари-Тута, также известная как наследственная моторная и сенсорная невропатия или малоберцовая мышечная атрофия, охватывает группу расстройств, которые влияют на периферические нервы. Периферические нервы лежат вне головного и спинного мозга. Нарушения, которые влияют на периферические нервы, называются периферическими невропатиями.
Симптомы болезни Шарко-Мари-Тута
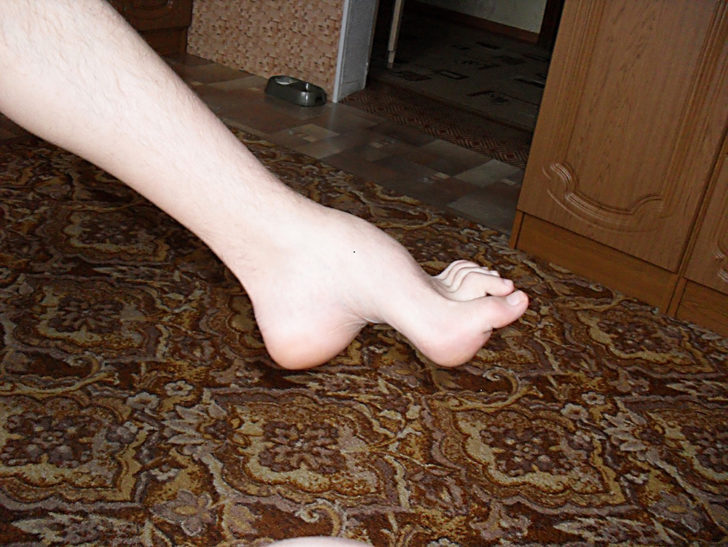
Невропатия влияет как на моторные, так и на сенсорные нервы. (Двигательные нервы заставляют мышцы сокращаться и возникает произвольная мышечная активность). Типичная особенность: слабость мышц стопы, что может привести к падению и нестабильной походке с частыми отклонениями или падениями. Из-за слабости мелких мышц в ногах характерны деформации стопы, похожие на высокие арки и молотки (состояние, при котором средний сустав пальца направлен вверх). Кроме того, голени ног могут приобретать форму «перевернутой бутылки шампанского» из-за потери мышечной массы. Позже у людей, страдающих этой болезнью могут возникать слабость и мышечная атрофия рук, что приводит к трудностям с выполнением тонких моторных навыков (координация небольших движений обычно в пальцах, руках, запястьях, ногах и языке).
Начало симптомов чаще всего происходит в подростковом возрасте или после совершеннолетия, но у некоторых люде симптомы развиваются к середине взрослой жизни. Тяжесть симптомов сильно различается у людей и даже среди членов семьи с этим заболеванием. Прогрессирование симптомов происходит постепенно. Боль может варьироваться от легкой до тяжелой, и некоторым людям могут потребоваться ходунки для опоры или другие ортопедические устройства для поддержания подвижности. Хотя в редких случаях у людей может развиваться слабость дыхательных мышц, болезнь Шарко-Мари-Тута не считается смертельным заболеванием, а люди с большинством форм данного заболевания имеют нормальную продолжительность жизни.
Причины болезни Шарко-Мари-Тута
Нервная клетка передает информацию в отдаленные объекты, посылая электрические сигналы по длинной тонкой части клетки, называемой аксоном. Чтобы увеличить скорость движения этих электрических сигналов, аксон изолирован миелиновой оболочкой, которая создается другим типом ячейки, называемой ячейкой Шванна. Миелин обертывает аксон вокруг и предотвращает потерю электрических сигналов. Без интактного аксона и миелиновой оболочки периферические нервные клетки неспособны активировать необходимые мышцы или передавать сенсорную информацию от конечностей обратно в мозг.
Болезнь Шарко-Мари-Тута вызвана мутациями в генах, которые продуцируют белки, участвующие в структуре и функции либо аксонов периферического нерва, либо миелиновой оболочки. Все мутации влияют на нормальную функцию периферических нервов. Следовательно, эти нервы медленно вырождаются и теряют способность передавать сигналы своим удаленным целям. Дегенерация двигательных нервов приводит к мышечной слабости и атрофии в конечностях (руки, ноги, предплечья или голени), а в некоторых случаях дегенерация сенсорных нервов приводит к уменьшению способности чувствовать тепло, холод и боль.
Генетические мутации при болезни обычно наследуются. Каждый из нас обычно имеет две копии каждого гена, один из которых унаследован от каждого родителя. Некоторые формы болезни Шарко-Мари-Тута наследуются аутосомно-доминантным способом, а это означает, что для причинения заболевания требуется только одна копия аномального гена. Другие формы заболевания наследуются аутосомно-рецессивным способом, а это означает, что обе копии аномального гена должны присутствовать для развития болезни. Другие формы Шарко-Мари-Тута наследуются по X-связанному способу, что означает, что аномальный ген расположен на Х-хромосоме. Х-хромосомы X и Y определяют пол индивидуума. Индивидуумами с двумя Х-хромосомами являются женщины и индивидуумы с одной Х и одна Y-хромосома - мужчины.
В редких случаях мутация гена, вызывающая болезнь, представляет собой новую мутацию, которая возникает спонтанно в генетическом материале человека и не передается через семейную историю.
Формы болезни Шарко-Мари-Тута
Существует много форм болезни Шарко-Мари-Тута, включая CMT1, CMT2, CMT3, CMT4 и CMTX. CMT1, вызванный аномалиями в оболочке миелина, имеет три основных типа. CMT1A является аутосомно-доминантным заболеванием, которое возникает в результате дублирования гена на хромосоме 17, которая несет инструкции по производству периферического белка миелина-22 (PMP-22). Белок PMP-22 является критическим компонентом миелиновой оболочки. Сверхэкспрессия этого гена вызывает ненормальность структуры и функции миелиновой оболочки. Пациенты испытывают слабость и атрофию мышц нижних конечностей, начиная с подросткового возраста; позже они испытывают слабость рук и сенсорную потерю. Интересно, что другая невропатия, отличная от CMT1A, называемая наследственной невропатией с предрасположенностью к параличу давления (HNPP), вызвана удалением одного из генов PMP-22. В этом случае аномально низкие уровни гена PMP-22 приводят к эпизодической рецидивирующей демиелинизирующей нейропатии. CMT1B является аутосомно-доминантным заболеванием, вызванным мутациями в гене, который несет инструкции по изготовлению нуклеотида миелина (P0), который является еще одним критическим компонентом миелиновой оболочки. Большинство из этих мутаций являются точечными мутациями, что означает, что ошибка встречается только в одной букве генетического кода ДНК. На сегодняшний день ученые выявили более 120 различных точечных мутаций в гене P0. В результате аномалий в P0 CMT1B вызывает симптомы, сходные с симптомами, обнаруженными в CMT1A. Менее распространенные CMT1C, CMT1D и CMT1E, которые также имеют симптомы, сходные с симптомами, обнаруженными в CMT1A, вызваны мутациями в генах LITAF, EGR2 и NEFL соответственно.
Болезнь Шарко-Мари-Тута является результатом аномалий в аксоне периферической нервной клетки, а не в оболочке миелина. Он менее распространен, чем CMT1. CMT2A, наиболее распространенная аксоновская форма CMT, вызвана мутациями в Mitofusin 2, белке, связанном с митохондриальным слиянием. CMT2A также был связан с мутациями в гене, который кодирует белок 1B-бета-члена семейства кинезина, но в других случаях он не реплицируется. Кинезины - это белки, которые действуют как двигатели, чтобы помочь обеспечить транспортировку материалов вдоль клетки. Другие менее распространенные формы CMT2 были недавно идентифицированы и связаны с различными генами: CMT2B (связанный с RAB7), CMT2D (GARS). CMT2E (NEFL), CMT2H (HSP27) и CMT2l (HSP22).
CMT3 или Dejerine-Sottas - тяжелая демиелинизирующая невропатия, которая начинается в младенчестве. У младенцев происходит тяжелая атрофия мышц, слабость и сенсорные проблемы. Это редкое расстройство может быть вызвано определенной точечной мутацией в гене P0 или точечной мутацией в гене PMP-22.
CMT4 включает несколько различных подтипов аутосомно-рецессивного демиелинизирующего двигателя и сенсорных невропатий. Каждый подтип невропатии вызван другой генетической мутацией, может влиять на конкретную этническую популяцию и производит различные физиологические или клинические характеристики. Лица с CMT4 обычно развивают симптомы слабости ног в детстве, а в подростковом возрасте они не могут ходить. Несколько генов были идентифицированы как вызывающие CMT4, включая GDAP1 (CMT4A), MTMR13 (CMT4B1), MTMR2 (CMT4B2), SH3TC2 (CMT4C), NDG1 (CMT4D), EGR2 (CMT4E), PRX (CMT4F), FDG4 (CMT4H) и фиг.4 (CMT4J).
CMTX возникает точечной мутацией гена connexin-32 на Х-хромосоме. Коннексин-32-белок экспрессируется в клетках Schwann-клеток, которые обертывают нервные аксоны, составляя один сегмент миелиновой оболочки. Этот белок может участвовать в связи с клеткой Шванна с аксоном. Мужчины, которые наследуют один мутированный ген от своих матерей, проявляют умеренные или тяжелые симптомы заболевания, начиная с позднего детства или подросткового возраста (Y-хромосома, которую самцы наследуют от своих отцов, не имеет ген коннексин-32). Женщины, которые наследуют один мутированный ген от одного родителя и одного нормального гена от другого родителя, могут проявлять мягкие симптомы в подростковом возрасте или позже или вообще не могут развиваться симптомы заболевания.
Диагностика болезни Шарко-Мари-Тута
Диагноз болезни Шарко-Мари-Тута начинается с изучения истории болезни, семейной истории и неврологического обследования. Больного будут спрашивать о характере и продолжительности его симптомов и о том, имеют ли другие члены семьи болезнь. Во время неврологического обследования врач будет искать признаки мышечной слабости в руках, ногах, руках и ногах человека, уменьшении объема мышц, уменьшении сухожильных рефлексов и сенсорных потерях. Врачи устанавливают признаки деформаций стопы, такие как высокие арки, молотки, перевернутые пятки или плоские стопы. Могут также присутствовать другие ортопедические проблемы, такие как умеренный сколиоз или дисплазия тазобедренного сустава. Специфическим признаком, который может быть обнаружен у людей с данным заболеванием, является увеличение нерва, которое может ощущаться или даже наблюдаться через кожу. Эти увеличенные нервы, называемые гипертрофическими нервами, вызваны аномально утолщенными миелиновыми оболочками.
При подозрении на болезнь Шарко-Мари-Тута, врачом может назначаться электродиагностические исследования. Это тестирование состоит из двух частей: исследования проводимости нервов и электромиографии (ЭМГ).
Во время исследований проводимости нерва электроды помещаются на кожу через периферический мотор или сенсорный нерв. Эти электроды создают небольшой электрический шок, который может вызвать легкий дискомфорт. Этот электрический импульс стимулирует сенсорные и двигательные нервы и предоставляет количественную информацию, которую врач может использовать для постановки диагноза. ЭМГ включает в себя вставку игольного электрода через кожу для измерения биоэлектрической активности мышц. Конкретные аномалии в показаниях означают дегенерацию аксона. ЭМГ может быть полезна для дальнейшего определения распределения и тяжести поражения периферических нервов.
Генетическое тестирование доступно для некоторых форм болезни Шарко-Мари-Тута, и результаты как правило достаточно для подтверждения диагноза. Кроме того, генетическое консультирование доступно для оказания помощи людям в понимании их состояния и планировании на будущее.
Если при электродиагностическом и генетическом тестировании результаты отрицательные, невролог может выполнить биопсию нерва для подтверждения диагноза. Биопсия нерва включает удаление небольшого куска периферического нерва через разрез в коже. Это чаще всего делается путем удаления кусочка нерва, который стекает по теленку ноги. Затем нерв исследуют под микроскопом. Лица с болезнью Шарко-Мари-Тута обычно обнаруживают признаки аномального миелинизации. В частности, можно увидеть «луковичные головки», которые представляют собой аксоны, окруженные слоями демиелинизирующих и ремиелинизирующих клеток Шванна. Люди с болезнью Шарко-Мари-Тута как правило обнаруживают признаки дегенерации аксона. Недавно биопсия кожи была использована для изучения немиелинизированных и миелиновых нервных волокон минимально инвазивным способом, но их клиническое применение при данном заболевании еще не установлено.
Лечение болезни Шарко-Мари-Тута
Лечение болезни Шарко-Мари-Тута не существует, но физическая терапия, профессиональная терапия, фигурные скобки и другие ортопедические устройства и даже ортопедическая хирургия могут помочь людям справиться с симптомами болезни, которые могут серьезно мешать нормальной жизни. Кроме того, лекарственные препараты могут назначаться для людей, имеющих сильные боли.
Физическая и профессиональная терапия болезни Шарко-Мари-Тута, включает в себя тренировку силы мышц, растяжение мышц и связок, тренировку выносливости и умеренные аэробные упражнения. Большинство терапевтов рекомендуют специализированную программу лечения, разработанную с одобрения врача человека для соответствия индивидуальным способностям и потребностям. Терапевты также предлагают начать раннюю программу лечения; усиление мышц может задержать или уменьшить атрофию мышц, поэтому силовые упражнения наиболее полезны, если они начинаются до того, как дегенерация нервов и мышечная слабость прогрессируют до места инвалидности.
Растяжка может предотвращать или уменьшать деформации суставов, которые являются результатом неравномерного растяжения мышц на костях. Упражнения для повышения выносливости или повышения выносливости помогут предотвратить усталость, которая возникает в результате повседневной деятельности, требующей силы и мобильности. Умеренная аэробная активность может помочь поддерживать сердечно-сосудистую систему и общее состояние здоровья. Большинство терапевтов рекомендуют упражнения с низким уровнем воздействия или бездействия, такие как езда на велосипеде или плавание, а не такие виды активности, как ходьба или бег трусцой, которые могут оказывать травмирующую нагрузку на хрупкие мышцы и суставы.
Многие пациенты с болезнью Шарко-Мари-Тута нуждаются в фиксаторах лодыжки и других ортопедических приспособлениях для поддержания повседневной мобильности и предотвращения травм. Держатели голеностопного сустава могут помочь предотвратить растяжение связок голеностопного сустава, обеспечивая поддержку и стабильность во время таких действий, как ходьба или подъем по лестнице. Высокие ботинки или ботинки могут также поддерживать слабые лодыжки. Большие пальцы могут помочь с ослаблением руки и потерей мелких моторных навыков. Вспомогательные устройства должны использоваться до появления инвалидности, поскольку устройства могут предотвратить мышечное напряжение и уменьшить мышечное ослабление. Некоторые люди с болезнью Шарко-Мари-Тута могут принять решение о проведении ортопедической хирургии для отмены деформаций стопы и суставов.
ЛАБОРАТОРНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ДЕТЕЙ ПЕРВЫХ 3 ЛЕТ ЖИЗНИ В НЕВРОЛОГИЧЕСКОЙ КЛИНИКЕНА ГЛАВНУЮ СТРАНИЦУ
Избранные статьи
